


Where did the SARS-CoV-2 Omicron variant come from?
Lessons learned from a scientific mystery

A recent compelling study suggests a possible origin of Omicron in rodents. Others indicate a connection with chronically infected patients. It is unclear whether the uncertainty will resolve anytime soon. And yet, one thing is certain: Western neglect of public health in developing countries is a historical problem with global ramifications.

Since the highly contagious SARS-CoV-2 variant B.1.1.529, now named Omicron, was discovered in Gauteng, South Africa in early November, it has been spreading around the world like wildfire. Even in countries that are currently still struggling with a strong delta wave, the Omicron variant is taking on an increasing proportion of all new infections. In other countries, Omicron has long been the practically sole cause of Covid-19.

After two years of the pandemic, the virus’ ability to surprise us with new variants seems hardly diminished. Where do these new variants come from and what makes Omicron so different from the previous ones? A team of scientists led by Weifeng Qian, professor of genetics at the University of the Chinese Academy of Sciences in Beijing, has now delivered a convincing hypothesis.
The animal origins of Omicron?
Soon after the discovery of the Omicron variant, scientists offered three potential origin scenarios; first a ‘hidden’ spread in a population group without genomic surveillance infrastructure, secondly a chronic infection in immunocompromised patients whose immune system cannot get rid of the virus for months, and thirdly an animal population infected by humans, which cultivated the virus and finally transmitted it back to humans as Omicron.
Professor Weifang Qian and his team took up this last point and searched the viral genomes of Omicron for clues of an animal host.
The scientists mainly focused on the many mutations that distinguish the first found Omicron sequences compared to the parent variant of SARS-CoV-2, and later Omicron genomes. More specifically, they examined whether there were certain patterns in the ‘mutation spectra’ between the earliest sequences (‘which may have just jumped out of the host animals’) and later Omicron genomes (‘all sequences obtained after November 15’), when the first Omicron wave spread in South Africa and accumulated more de novo mutations.
Mutation spectra describe a nucleotide preference of de novo mutations. Since RNA consists of 4 nucleotide bases (A, C, G, U), there is a theoretical ~33% chance that e.g. an A becomes a C. In practice, however, it is more complicated, since mutations underlie many internal and external processes that entail characteristic chemical consequences. For example, a ‘C to U transition’ is often indicative of RNA editing processes due to cytosine deamination. This deamination can in turn be triggered by UV light , for example, so mutational signature can be a roundabout way of identifying environmental factors shaping mutations.
A different set of mutational preferences were also found in relation to tobacco smoking. In cancer research, there has been an increasing search for these ‘mutation profiles’ or ‘mutational signatures’ as quasi-molecular fingerprints for cancer-causing substances and environmental influences for years. But what about viruses?
In a previous publication, Weifen Qian’s team was able to show that de novo mutations in SARS-CoV-2 are very strongly determined by the cellular environment of the host organism and are not so much dependent on the virus (and its replication machinery) itself. To their surprise, they found a mutation spectrum in early Omicron sequences that is neither comparable to later Omicron spectra nor to the general distribution of de novo mutations in SARS-CoV-2. However, late Omicron sequences coincided with the general mutation preferences of SARS-CoV-2, thus illustrating a selection in humans.
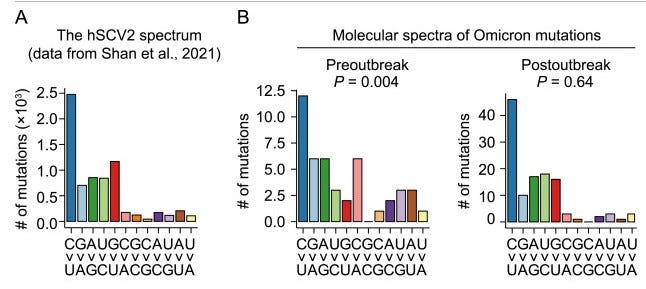
This statistical deviation is strange at first but is by no means a conclusive argument for an animal host. The scientists had to dig deeper. So the next step was to turn to coronaviruses isolated from different mammalian species. They created phylogenetic family trees for each animal family in order to then draw conclusions about acquired de novo mutations and finally generate mutation spectra for individual animal backgrounds. The wide range of different mutation spectra was then aggregated into one dataset and visualized using computer-assisted clustering methods. The scientists were able to determine that mutation spectra from the same host animal background matched one another much better than spectra from other host animals. Using this rough ‘navigation map’ of the mutation spectra, the scientists were then able to show that coronaviruses in rodents (tested: mice) have very similar mutation patterns to the early (but not later) Omicron genomes.
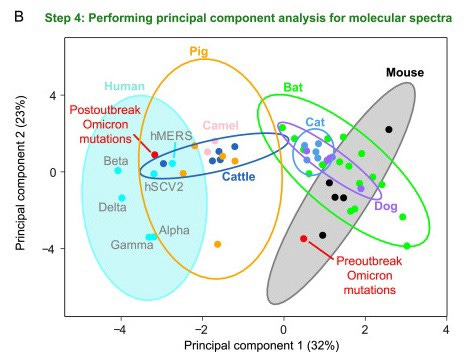
”We are fairly certain that Omicron did not evolve in humans,” explains Weifang Qian when asked. He thinks mice are a good candidate, but he can’t conclusively confirm the exact host species because the current resolution of the mutation spectra isn’t high enough for that.
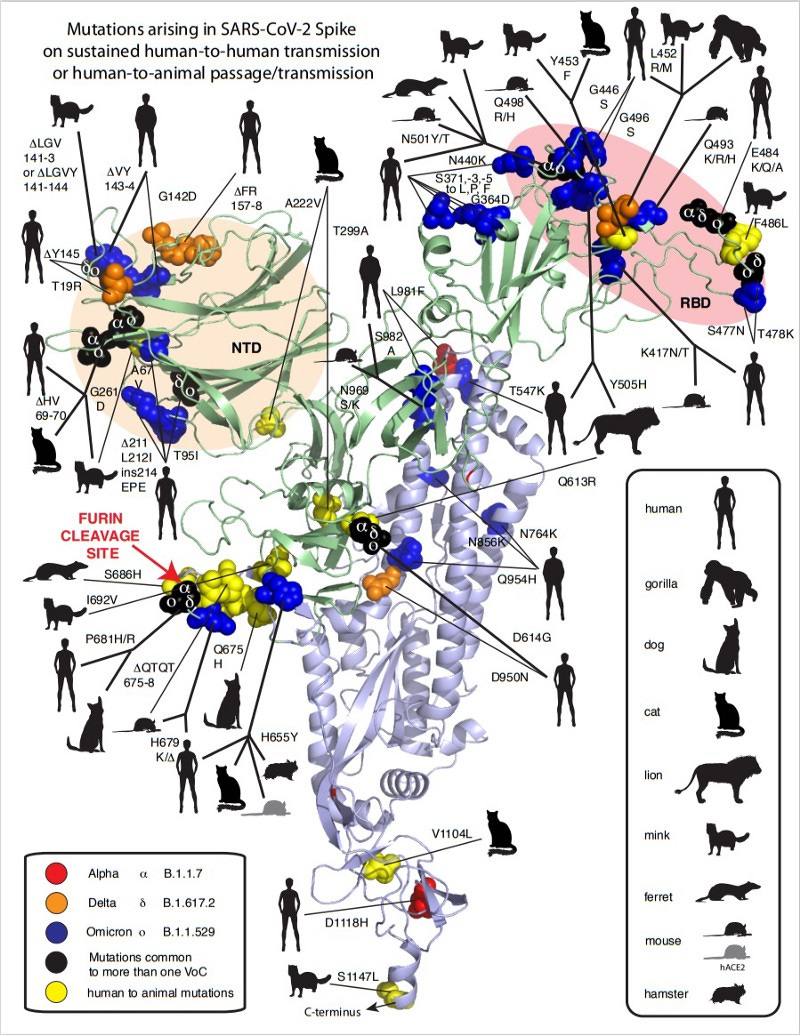
However, Qian is not alone in his belief in rodents. Other scientists have observed that characteristic mutations such as Q498R/H or N501Y in the Omicron viral spike protein increase rodent tropism. In contrast to Omicron (but also e.g. the Alpha variant from Great Britain), SARS-CoV-2 and the Delta variant cannot infect mice and rats. A recent study also confirms that these characteristic mutations have dramatically increased binding affinity to rat cell-entry receptors. Other scientists found traces of these characteristic mutations in New York City sewage surveys months before Omicron existed. Although they could not trace the origins of these sequences, the scientists suspected an animal origin, potentially rats. Follow-up experiments showed that these cryptic lineages with omicron-like mutations were capable of efficiently transducing cells with mice and rat ACE2. Could this be a sign of convergent evolution?
Rats are well known in history as disease vectors, but how exactly do they fit into Omicron’s picture?
Of rats and men
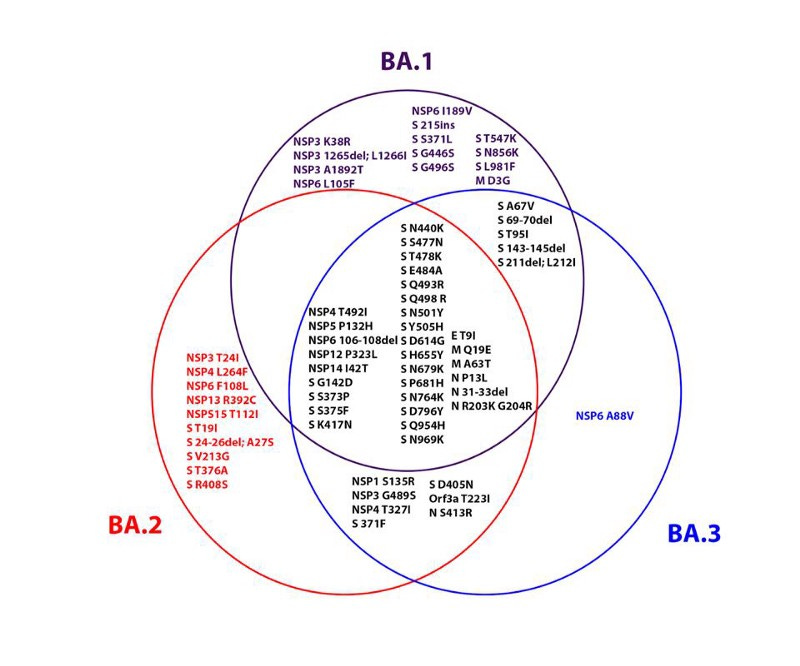
We do not have definitive epidemiological data on the origins of Omicron. The first confirmed Omicron cases were retrospectively dated November 9 by the WHO; however, due to the S-gene dropout in PCR tests, scientists suspect that by this time Omicron had already replaced Delta as the dominant variant. A recently published Nature study from South Africa also shows that three evolutionarily related but independent Omicron lineages (BA.1, BA.2, BA.3) were rampant in Gauteng Province, South Africa by the end of November. Modeling puts the start time for human-to-human transmission chains of Omicron in early October.
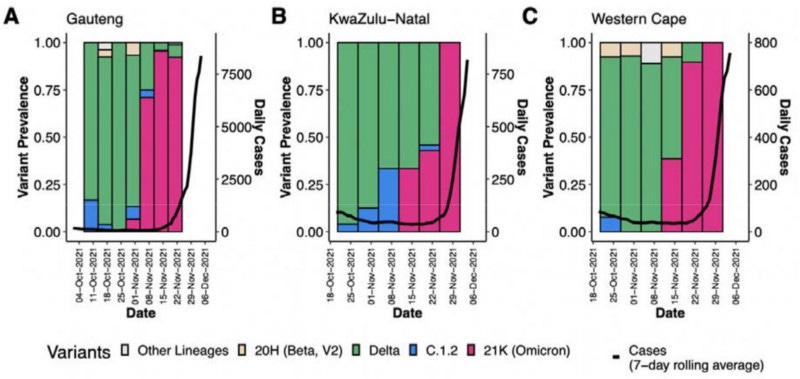
It is difficult to prove with certainty whether the province of Gauteng, with the city of Johannesburg, really represented the site of the zoonotic jump or simply an epicenter of the infections.
If the SARS-CoV-2 virus has shown us one thing, it is that it is capable of jumping into all kinds of animals. From exotic raccoon dogs, civet cats or armadillos to mink, ferrets or deer, whether pets, zoo animals or wild animals, our human incompetence to contain the virus has now affected many other animals with which we come into contact. Zoonotic jumps between animals and humans require one thing above all: many opportunities. Conversely, this means that the probability of a reverse zoonotic from animals to humans is given when a correspondingly large animal population first circulates the virus and humans are in frequent contact with these animals. Rats in dense cities seem predestined for this.
Belgian scientists also pursued this idea, who tested rats for potential infections after a SARS-CoV-2 wave in the city of Antwerp in November 2020, but found no evidence of this. Another study from Hong Kong in February 2021 also found no direct evidence of acute infections, but one of the 213 animals tested had specific antibodies against SARS-CoV-2, which was not due to cross-reaction with other circulating alphacoronaviruses. At the time of this study (and up to now), there were relatively few SC2 infections overall in Hong Kong and it is questionable whether there was ever enough opportunity for spillover into rats.
However, in stark contrast to the well-groomed Antwerp or pandemic-resistant Hong Kong, the situation in other cities looks much more dramatic. In Johannesburg, for example, a third of all residents were infected in the first wave in mid-2020, right about the time where phylogenetic analysis puts the last common ancestor of Omicron. Additionally, Johannesburg has been mired in a public health crisis triggered by rats for decades. Studies found rat infestations in over 50% of all households; Households in poorer townships in particular, where sewage treatment and waste disposal have historically been problematic, report that rats have caused dramatic damage to residents.

Could such an environment set the right conditions for zoonotic leaps?
Weifeng Qian thinks similarly.
“I think it’s entirely possible that rats could also have been a likely host for Omicron to accumulate mutations there. Originally, our study intended to use the broader title ‘rodents’, but we didn’t really have any evidence for any rodents other than mice”.
Dr. Jacqueline Weyer from the Centre for Emerging Zoonotic and Parasitic Diseases (CEZPD) in South Africa says that there are no surveillance programs in rodents for SARS-CoV-2, because these lack a scientific rationale at this point. Human-to-human transmissions are the predominant route for SC2 in South Africa. While there are incidental infections of animals, they have not been aware of any sustained transmission of SC2 in South African animals yet. While speculative in South Africa, an infection study in deer mice done in North America already showed that in principle, there is a clear potential for sustained rodent-to-rodent transmission of SARS-CoV-2.
How viral transmission from rats back into humans might work can be seen in other rodent-derived roboviruses, e.g. hantavirus or arenaviruses, which are normally transmitted through rat excretions. Zoonotic jumps from these roboviruses to humans occur sporadically, but do not usually result in further human-to-human transmission. It is not yet clear whether and through which routes the respiratory SARS-CoV-2 can make its way back from rat to humans.
Lourens Swanepoel from the University of Venda thinks there are ‘definitively enough rodent-human encounters to provide opportunity’ of zoonotic transfers, but his work focuses mostly on rural areas where native murine species, not commensal city rats, are prevalent.
Professor Amanda Bastos from the University of Pretoria, South Africa, has worked on the zoonotic risks posed by rats for years. She says that depending on the pathogen, several transmission routes with rats are possible. From rat droppings to shared food sources to direct attacks. She further pointed out that media reporting in South Africa suggested rat infestation increased substantially during the pandemic.
“Most likely due to the drop on pest control efforts during covid lockdowns”
Yet she doesn’t have any hard numbers on that.
How present the danger really is will therefore require further studies. That these costly genetic surveillance programs in high-risk cities have not happened yet is equally understandable as it is worrying.
The mysterious early epidemiology of the Omicron lineages
A zoonotic origin hypothesis would definitely find epidemiological support. From the beginning, Omicron already had several genetically separated lineages (named BA.1, BA.2 and BA.3). The fact that these Omicron cousin lineages are so genetically distinct is curious since the time between first human Omicron infections and the almost simultaneous emergence of BA.1 and BA.2 hardly explains such strong diversification from one another. The phylogeny suggests that either this segregation of the highly contagious Omicron lineages happened unnoticed in humans months ago, or we are observing multiple jumps from an infected animal reservoir.
Spyros Lytras, a PhD student at Glasgow’s Center for Viral Research, sees the various Omicron sublineages emerging almost simultaneously as harder to reconcile with a single chronic infection explanation. The fact that Omicron was first found in South Africa, which has an excellent sequencing infrastructure and is arguably the main travel hub for other parts of Africa without genomic observation, does not surprise him. Coupled with the high unvaccinated rate in Africa, he still thinks an ‘undetected’ outbreak is just as consistent with the emergence of Omicron sub-lineages as multiple zoonotic jumps back into humans from an animal reservoir.
“What’s encouraging about a potential animal origin is the fact that we could still find Omicron ‘pre-sequences”
he says. So at least theoretically, a conclusive answer to this question might be out there. However, we have to be quick, because at some point Omicron, which constantly jumps from infected humans to animals, will also blur this background.
An animal reservoir also provides a good explanation for Omicron’s unusual properties, including a different molecular cell entry mechanism. A selection study of Omicron mutations, led by Prof. Sergei Pond of Temple University in Philadelphia, indicates that the many unusual mutations in Omicrons alter the biological function of the spike protein so profoundly that they reflect an antigenic shift rather than the more gradual antigenic drift. This is quite curious.
In influenza viruses, antigenic drift mechanisms are often attributed to spread in humans, while the more dramatic antigenic shifts are more likely attributed to animal reservoirs or recombination with other viruses.
However, the same selection study also showed that Omicron mutations, while individually less fit, seem to be adaptive in bulk, which means they must have arisen almost simultaneously. How this came about, Sergei Pond is less sure, but he believes that a chronic infection in humans must have played an important role at some point. He is by far not alone.
So what about immunocompromised patients?
A good amount of evidence might point towards a potential zoonotic origin of the Omicron variant. Infection studies in rats, as well as genomic surveillance studies in the relevant animal populations, could provide further clarity.
However, it is another potential origin of Omicron which, based on the current evidence, might in fact be more capable of explaining how certain features of Omicron could have evolved.
A recently published case study of a SARS-CoV-2 infected AIDS patient attracted attention. This patient tested positive for SARS-CoV-2 for months without getting rid of the virus and often had to be hospitalized due to health problems. The samples taken allowed scientists to retrospectively track the evolution of the SARS-CoV-2 virus. Over time, the scientists noted an accumulation of mutations that helped the virus evade both contagious and vaccine-induced neutralizing antibodies. Some of these ‘immune escape’ mutations are also present in Omicron. Other case studies in HIV patients since then have also consistently found Omicron-like mutations or an accumulation of mutations that may bypass the neutralizing effects of antibodies. Furthermore, a structural study showed that the Spike protein in Omicron evolved towards a more closed physical configuration, indicating to researchers that prolonged exposure to an immune system likely was driving this change in structure. Additionally, the ratio of non-synonymous to synonymous (dN/dS) mutations, a useful metric to assess selection pressures, also points towards a very strong positive selection of Omicrons mutations.

It is exactly these findings that make it difficult for Prof. Andrew Rambaut from the University of Edinburgh to imagine any non-human host, or truly any transmission between populations, are at all capable of producing these mutations. He points out that the viral diversity in a single chronically infected individual is way higher than in a population because these viral genomes don’t get ‘trimmed’ by purifying selection between transmissions. Other studies agree that transmission bottlenecks between hosts are narrow for SC2, even in superspreading events, and dramatically slow down viral evolution. Some studies even suggest that peeking into chronically infected patients can give a glimpse of how viruses might evolve in the future.
Andrew Rambaut thinks a chronically infected patient is the only cellular environment that would apply such strong and prolonged selection pressures which can drive all of these immune-evasive mutations we observe in Omicron. He also points out that SARS-CoV-2 barely changed when it jumped into Minks in Denmark and the Netherlands, and it has since been shown that the few mutations acquired in this animal reservoir did not increase viral fitness. Omicron’s high mutational diversity is unlike anything we have observed in SC2 animal reservoirs until today.
However, Christian Drosten from the Charité Hospital Berlin sees this high mutational diversity as a potential sign against a ‘chronic’ origin of Omicron, since the additional burden of escape mutations is usually accompanied by other changes that can impair the ability to be infected, as is the case, for example, with chronic infections with influenza viruses. ‘These [chronic] viruses show reduced fitness in the real world outside,’ he said in a Science Magazine interview. The chronic infection hypothesis would be somewhat less consistent with the increased contagiousness of the various Omicron lineages, but it is by far not its only problem.
The almost simultaneous outbreak of multiple related Omicron sub-lineages is hard to explain having an origin in a single human host. Because these three sub-lineages diverged early, being basically distant cousins of each other, the chronically infected patient must have held these three competing lineages in the same body, without one out-competing the other two; a kind of dynamic stalemate for months where three distinct viral sublineages evolved against the human immune system and each other in a single body. And while all this internal competition and strong selection pressure were going on, both BA.1 and BA.2 seemed to have found unique ways to also increase transmission capabilities to better infect other hosts.
While mindbendingly unlikely, there are scientifically established precedents of these dynamics with other viral infections, for example, HIV superinfections. These superinfected patients harboring multiple lines can also transmit more than one viral lineage to others. Andrew Rambaut sees in the early evidence that BA.2 sub-lineage is now substituting BA.1 in multiple countries as a potential feature of these co-evolved dynamics. So far, we have not observed these proposed ‘stalemate’ SARS-CoV-2 lineages dynamics in HIV patients or anywhere else.
Similar to the suggested surveillance in animal reservoirs, more studies and observations in chronically infected patients may well confirm this hypothesis eventually.
Finally, we must keep in mind that all proposed hypotheses still suffer from incomplete data.
Therefore, even a ‘conventional’ selection of Omicron in unobserved population groups, especially rural or low-income social groups without access to medical facilities, cannot be completely ruled out at this point in time. It is questionable if such groups could ever exert the necessary selection pressure to allow an SC2 mutant variant such as Omicron to emerge in a relatively short time. Furthermore, the wildfire-like spread of the various Omicron sub-lineages shortly after their discovery in South Africa leaves it open whether they really could have circulated silently in other African countries for months without being noticed.
Whether HIV patients or unobserved groups, pure selection in humans is difficult to reconcile with the non-human ‘mutation spectra’ of early Omicron sequences found by Wenfeng Qian. On the other hand, can a viral stay in an animal reservoir really account for the sheer number of these immune-escape mutations given transmission bottlenecks? And what about the higher transmission capacity and simultaneous emergence of multiple sub-lineages? At this point, even a combination of various human and animal factors cannot be ruled out either.

Precisely because of these ambiguities, many scientists are still reluctant to attempt a definitive answer to the question of the origin of Omicron. Kristian Andersen, an infectious diseases researcher at the Scripps Research Institute in California, used to lean towards a ‘reverse zoonosis’ explanation for Omicron but finds the emerging data on strong positive selection in HIV patients to be the most compelling hypothesis at the moment. Everybody I’ve talked to agrees it is too early to completely close the door on any scenario.
A lot might still change when new data comes in. After all, Omicron is only a few months old and there will be more to learn in the future when looking deeper into potential animal reservoirs and chronically infected patients. Today, it might still be a mystery, but we can rest assured that some of our brightest minds are working on it.
This pointing towards the future is usually where an article like this would end. However, I can not help but wonder what lessons Omicron is truly teaching us already today?
Lessons learned?
As so often in this pandemic of uncertainties, there is no absolute clarity about the origins of Omicron, only good hypotheses. However, a common thread can be identified in all of the proposed origin scenarios: What they all have in common is the decades of neglect of public health in developing countries.
Omicron teaches us that urban rat plagues, the AIDS pandemic, or the lack of access to health care in Africa are unfortunately more than just a local problem in a networked world. As we’ve painfully learned with viruses, any local or remote gap in attention can quickly turn into a global catastrophe.
Many reports rightly condemn the unequal distribution of SARS-CoV-2 vaccines worldwide. At the moment, more than 1 billion people in Africa are still waiting for their first dose, while in Europe and North America many booster campaigns are failing, not because of the availability of active ingredients, but because of the willingness to vaccinate.
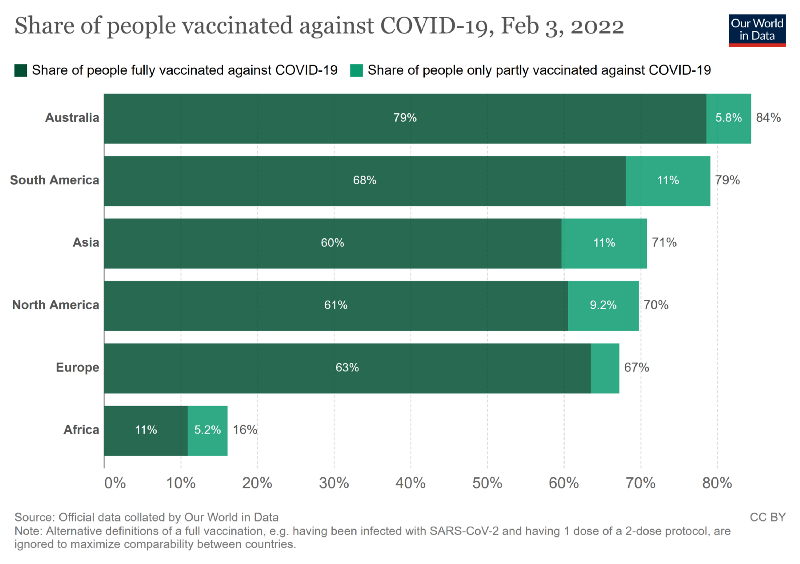
Vaccination gaps are also a dramatic issue for us in developed countries, as they give the virus more chances of making people seriously ill and also collecting critical mutations. It is time to get our act together. Governments rightfully call on the citizens to close these ‘vaccination gaps’ and increasingly consider vaccine mandates.
However slow we are to fill vaccination gaps here at home, we feel even less need to act in fixing other nations’ ‘public health gaps’, although by now it should be clear to everyone that wildlife trade and factory farming are hardly risk-free practices.
When will we learn that our health is inevitably linked to the public health of other countries?
The emergence of Omicron, like the pandemic itself, shows that we need to think of ‘public’ health broader than the national management of a pathogen. Rat plagues and AIDS have raged in African countries for decades. South Africa’s superior genomic surveillance system allowed the world to be warned about Omicron. But this is only one side of the coin. Real pandemic preparedness takes a responsible global civilization to support African nations in creating living circumstances that drastically reduce the risk of evolving such viral pathogens in the first place. So far, we’ve not even managed the most basic of interventions, a temporary lift of patent restrictions on the Covid vaccines, so other countries might have a chance to catch up on vaccination. We need deeper reforms fast.
SARS-CoV-2 will become endemic. We will probably never be able to completely avoid zoonotic spillovers or prevent viral evolution. However, that doesn’t mean we’re powerless. We can and should minimize unnecessary or particularly risky circumstances, wherever they happen in the world. Given our global neglect, the emergence of variants like Omicron was probably inevitable in today’s world.
In tomorrow’s, it need not be.
If we don’t want to keep repeating the same mistakes, the time to act is now. Omicron likely won’t be the last dangerous SARS-CoV-2 variant we have to reckon with. In a connected world full of unwise practices, animal exploitation, and searing public health gaps, other zoonotic pandemics are likely to follow SARS-CoV-2’s trajectory eventually.
I know that for all the uncertainty which has been surrounding us, this might be the one sobering clarity science can give us today.
References:
Kupferschmid Kai, Science, 2021
Wei C. et al., Journal of Genetics and Genomics, 2021
Barak Y. et al., J. Biol. Chem, 1995
Chen Z. et al., BMC Cancer, 2020
Shan K. et al., The Innovation, 2021
Shuai H. et al., The Lancet, 2021
Bate N. et al., biorxiv, 2021
Smyth DS. et al., Nature, 2022
Viana R. et al., Nature, 2022
Colombo VC. et al., Transbound Emerg Dis., 2021
Miot E. et al., Emerging Infectious Diseases, 2022
George JA. et al., SAMJ, 2021
Jassat W. et al., International Journal of Environmental Health Research, 2013
Chelule PK. et al., Int J Environ Res Public Health, 2021
Fagre A. et al., PLoS Pathog., 2021
Willet BJ. et al., medrxiv, 2022
Martin DP. et al., biorxiv, 2022
Dowdle WR. et al., Bull Pan Am Health Organ, 1976
Cele S. et al., Cell Host & Microbe, 2022
Riddell AC. et al., medrxiv, 2022
Maponga TG. et al., SSRN, 2022
Gobeil SMC. et al., biorxiv, 2022
Kryazhimskiy S. et al., PLoS Genetics, 2008
Morales AC. et al., Genome Biology and Evolution, 2021
Braun KM. et al., PLoS Pathogens, 2021
Hannon WW. et al., biorxiv, 2022
Kemp SA. et al., Nature, 2021
Oreshkova N. et al., Euro Surveill., 2021
Zhu J. et al., Cell reports, 2022
Gao Y. et al., Front Med., 2017
Novitsky V. et al., PLoS One, 2016